Fenylketonúria
Marian Grosser vyštudoval humánnu medicínu v Mníchove. Lekár, ktorý sa zaujímal o veľa vecí, sa navyše odvážil urobiť niekoľko vzrušujúcich obchádzok: štúdium filozofie a dejín umenia, práca v rozhlase a nakoniec aj pre doktora.
Viac o expertoch na Všetok obsahu kontrolujú lekárski novinári.Fenylketonúria (PKU) je vrodené, dedičné ochorenie metabolizmu bielkovín. Zabraňuje odbúravaniu aminokyseliny fenylalanínu. To sa hromadí v tele a narúša vývoj mozgu dieťaťa. Ak sa fenylketonúria nelieči, vedie k ťažkému mentálnemu postihnutiu. Včasnou terapiou však môžu pacienti viesť normálny život. Tu sa dozviete všetko o fenylketonúrii!
Kódy ICD pre túto chorobu: Kódy ICD sú medzinárodne uznávané kódy pre lekárske diagnózy. Môžu sa nachádzať napríklad v listoch lekárov alebo na potvrdeniach o práceneschopnosti. E70
Fenylketonúria: popis
Fenylketonúria (PKU) je dedičné metabolické ochorenie, ktoré existuje od narodenia a interferuje s rozkladom esenciálnej aminokyseliny fenylalanínu. Aminokyseliny sú základnými stavebnými kameňmi bielkovín a teda životne dôležitých metabolických zložiek. Niektoré z nich sa do tela môžu dostať iba prostredníctvom potravy, organizmus si ich nevie sám vyrobiť. Takéto aminokyseliny sa nazývajú esenciálne.
Čo sa stane s fenylketonúriou?
Aminokyseliny za normálnych okolností podliehajú rovnováhe medzi absorpciou / hromadením a rozkladom, takže je k dispozícii vždy toľko, koľko telo potrebuje. V prípade rôznych aminokyselín môže nedostatok alebo prebytok spôsobiť značné škody a spôsobiť rôzne príznaky.
Pri takzvanej klasickej PKU je účinok fenylalanínhydroxylázy (PAH) obmedzený alebo dokonca úplne chýba. V dôsledku nedostatku PAH sa fenylalanín v tele stále viac hromadí. Príliš vysoká koncentrácia fenylalanínu značne narúša vývoj mozgu a v ranom štádiu vedie u mladých pacientov k mentálnemu postihnutiu.
Pretože pri chorobe nie je možné normálne odbúravanie fenylalanínu, vytvárajú sa ďalšie produkty rozpadu, takzvané fenylketóny. Vylučujú sa močom a sú zodpovedné za názov choroby.
Atypická fenylketonúria
Aj pri atypických formách fenylketonúrie je odbúravanie fenylalanínu narušené. Príčinou však nie je chyba PAH. Namiesto toho je obmedzená funkcia koenzýmu, tetrahydrobiopterínu (BH4). Nepriamo sa podieľa na odbúravaní fenylalanínu, pretože PAH potrebuje BH4 na premenu fenylalanínu na tyrozín.
Pretože BH4 je tiež dôležitý pre produkciu nosných látok dopamínu a serotonínu, atypická fenylketonúria s nedostatkom BH4 je zvyčajne komplikovanejšia ako klasická forma.
Koho postihuje fenylketonúria?
PKU je jednou z najčastejších vrodených metabolických chorôb. Odhaduje sa, že ho vyvinie približne jeden zo 7 000 novorodencov na celom svete bez rozdielu medzi dievčatami a chlapcami. Keďže ide o dedičné ochorenie, často je postihnutých niekoľko členov rodiny.
Fenyketonúria: príznaky
Deti s fenylketonúriou najskôr nevykazujú žiadne príznaky ochorenia. Prvé problémy nastávajú až vo štvrtom až šiestom mesiaci života, ak choroba ešte nebola rozpoznaná a liečená. Predovšetkým narušené dozrievanie mozgu spôsobuje časom obrovské komplikácie. Príznaky neliečenej PKU zahŕňajú:
- silný duševný deficit. Poškodenie mozgu prebieha počas puberty a potom stagnuje. Postihnuté deti sú potom spravidla ťažko mentálne postihnuté.
- Záchvaty záchvatov (epilepsia). Z dôvodu poškodenia sú nervové bunky mozgu obzvlášť citlivé a preexponované. Výsledkom sú časté epileptické záchvaty.
- motorické postihnutie. Nielen mozgové bunky, ale aj svaly pacienta môžu byť prebudené. Preto sa často stáva napätým (spasticita), čo vedie k rôznym pohybovým poruchám.
- Poruchy správania. Niektoré deti s fenylketonúriou sú hyperaktívne a neobvykle agresívne a častejšie sú aj výbuchy hnevu.
- malá hlava (mikrocefália). Pretože sa mozog pacienta nevyvíja správne, zaostáva aj rast hlavy. Malý obvod hlavy v porovnaní s rovesníkmi je obzvlášť výrazný u starších detí.
- znateľný zápach. PKU produkuje určité produkty rozkladu fenylalanínu, ktoré páchnu podobne ako myšie exkrementy. Tieto látky sa vylučujú hlavne močom, ale čiastočne aj kožou.
- kožné zmeny podobné ekzému
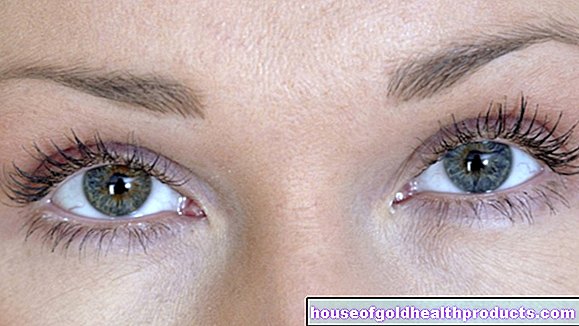
Pretože produkcia pigmentu melanínu je narušená aj vo fenylketonúrii, mnoho trpiacich má veľmi svetlú pokožku citlivú na slnko a bielo-blond vlasy. Dúhovka očí je tiež svetlo modrá až priehľadná a umožňuje presvitanie červenkastého očného pozadia.
Príznaky PKU sa líšia závažnosťou od človeka k človeku. Hlavným dôvodom je to, že aktivita fenylalanínhydroxylázy (PAH) je u každého pacienta obmedzená inak. Niektoré majú ešte určitú zvyškovú aktivitu, takže sa v organizme nahromadí menej fenylalanínu. Iní nevykazujú žiadnu enzýmovú aktivitu - choroba postupuje zodpovedajúcim spôsobom rýchlejšie a vážnejšie.
Fenylketonúria: príčiny a rizikové faktory
Fenylketonúria je dedičné ochorenie. Teraz je známych množstvo genetických mutácií, ktoré vedú k defektu PAH. Typ mutácie určuje, do akej miery je obmedzený rozklad fenylalanínu.
PKU je dedične recesívna, čo znamená, že človek môže byť nosičom zmeneného génu bez toho, aby sa u neho choroba rozvinula. Rovnako tak ľudia s fenylketonúriou môžu splodiť zdravé deti.
Iba vtedy, ak majú obaja rodičia mutácie v genetickej výbave, existuje určitá pravdepodobnosť, že sa u ich potomstva vyvinie fenylketonúria. Ak sú rodičia nielen nositeľmi génov, ale majú aj samotnú PKU, vyvinú ju všetky deti spoločne.
Fenylketonúria: vyšetrenia a diagnostika
Pretože závažným následkom fenylketonúrie možno predísť včasným začatím liečby, je obzvlášť dôležité odhaliť ochorenie čo najskôr. V Nemecku sú deti vyšetrované na rôzne vrodené choroby vrátane PKU v rámci všeobecného vyšetrenia (skríning novorodencov) tretí deň po narodení.
Tandemová hmotnostná spektrometria
Mnoho vrodených metabolických porúch je dnes diagnostikovaných pomocou takzvanej tandemovej hmotnostnej spektrometrie. Umožňuje rýchle a ľahké vyšetrenie krvi novorodenca. Okrem fenylketonúrie môžu lekári v priebehu niekoľkých minút odhaliť viac ako 20 ďalších chorôb.
Guthrieho test
Guthrieho test pomenovaný po svojom vynálezcovi umožňuje diagnostikovať aj PKU. Za týmto účelom sa dieťaťu odoberie malé množstvo krvi z päty a nanesie sa na kus filtračného papiera. V laboratóriu potom môžete určiť, či je koncentrácia fenylalanínu zvýšená.
Guthrieho test bol zavedený v 60. rokoch minulého storočia a je už dlho štandardnou metódou diagnostiky fenylketonúrie. V porovnaní s tandemovou hmotnostnou spektrometriou má však svoje nevýhody. Guthrieho test prináša výsledok až po piatich dňoch, ktorý je tiež náchylný na chyby. Výsledky napríklad sfalšujú faktory ako diéta dieťaťa alebo možná antibiotická terapia. V niektorých krajinách sa stále používa Guthrieho test, v Nemecku sa už spravidla nepoužíva.
Ďalšie testy
Ak skríning novorodenca odhalí podozrenie na fenylketonúriu, nasleduje ďalšie vyšetrenie na potvrdenie. To sa dá použiť aj na stanovenie presnej koncentrácie fenylalanínu v krvi.
Nakoniec je dôležité rozlíšiť, či ide o typickú (klasickú) alebo atypickú PKU. Na tento účel sú k dispozícii aj špeciálne testy, ako napríklad stresový test tetrahydrobiopterínu. Toto rozlíšenie je dôležité, pretože s atypickou fenylketonúriou sa zaobchádza inak ako s klasickou formou.
Vyšetrenie plodovej vody
Fenylketonúriu je možné diagnostikovať už počas tehotenstva (prenatálna diagnostika). Za týmto účelom sa odoberie malé množstvo plodovej vody z plodového vaku matky a skúmajú sa bunky nenarodeného dieťaťa v ňom obsiahnuté. Týmto spôsobom je možné identifikovať akékoľvek genetické chyby, ktoré spôsobujú PKU.
Pri skríningu novorodencov sa však fenylketonúria zvyčajne objaví dostatočne skoro na liečbu. Pretože test na plodovú vodu je vždy spojený s určitým rizikom, jeho použitie na diagnostiku PKU zvyčajne nie je užitočné.
Fenylketonúria: Liečba
Existuje iba jeden spôsob, ako pôsobiť proti nadbytku fenylalanínu v PKU: Postihnuté deti musia dodržiavať špeciálnu diétu, aby mohli spolu s jedlom prijímať čo najmenej fenylalanínu. Potrebujú tiež určité potravinové doplnky, ktoré nahradia látky, ktoré by sa u zdravých detí tvorili z fenylalanínu.
Terapia musí začať skôr, ako sa objavia prvé príznaky vývojovej poruchy, t.j. počas prvých dvoch mesiacov života. Poškodenie mozgu, ktoré už nastalo, nie je možné vrátiť späť.
Nutričná terapia s diétou PKU
Všetky prírodné proteíny pozostávajú z asi piatich percent aminokyseliny fenylalanínu. Väčšina potravín ich obsahuje oveľa viac, ako telo potrebuje. Pre zdravého človeka to nie je problém, pretože sa rozkladá a vylučuje prebytočné množstvo fenylalanínu.
Pacienti s fenylketonúriou sa naopak musia z väčšej časti zaobísť bez prírodných bielkovín. Priemyselne vyrábané špeciálne výrobky nahrádzajú chýbajúce zložky potravín. Na jednej strane chce človek zabrániť prebytku fenylalanínu, na strane druhej by mal byť pacient zásobený látkami, ktoré by mu inak chýbali, ako napríklad tyrozín, ktorý sa tvorí z fenylalanínu.
Cieľom diéty PKU však nie je úplne zastaviť absorpciu fenylalanínu. Pretože organizmus potrebuje určité množstvo aminokyseliny na dôležité metabolické procesy. Dosiahnutie správnej koncentrácie predstavuje pre lekárov a pacientov veľkú výzvu a vyžaduje si veľkú disciplínu.
Diétu je preto potrebné dodržiavať celý život, ale obzvlášť prísne do šiestich rokov. Pretože sa mozog až do tohto veku vyvíja veľmi silne, a preto je obzvlášť náchylný na poškodenie. V dospelosti vysoké koncentrácie fenylalanínu nespôsobujú poškodenie mozgu ako u detí. Môžu však byť spúšťačom ďalších neurologických ťažkostí, ako je slabá koncentrácia alebo spomalené reakcie.
Diétna liečba fenylketonúrie by mala začať v špecializovanom centre metabolických chorôb. Pretože nie je možné na každej klinike poučiť rodičov o diéte. Najlepšie sa to dá dosiahnuť pomocou diétneho poradenstva, ktoré ukazuje, ako držať diétu a pravidelne monitorovať hladinu fenylalanínu v krvi.
Liečba atypickej fenylketonúrie
V atypických formách PKU je umelo nahradený chýbajúci koenzým BH4 a určité posolské látky, ako je dopamín a serotonín. V niektorých prípadoch musí byť pacient aj na diéte s nízkym obsahom fenylalanínu.
Fenylketonúria počas tehotenstva
Ak ste tehotná a máte sama fenylketonúriu, mali by ste zvážiť nasledujúce:
- Dodržujte svoju diétu obzvlášť prísne!
- Nechajte si často kontrolovať hodnoty fenylalanínu v krvi. Pretože koncentrácie fenylalanínu u nenarodeného dieťaťa sú asi dvakrát vyššie ako u tehotnej ženy. Nenarodenému dieťaťu môžu okrem mozgu poškodiť aj srdce a oči. Malformácie kostry dieťaťa môžu tiež vzniknúť z vysokých hladín fenylalanínu počas tehotenstva.
- Aby sa vylúčilo poškodenie mozgu dieťaťa na začiatku tehotenstva, ženy s fenylketonúriou by mali tehotenstvo starostlivo naplánovať a už od začiatku vykonávať preventívne opatrenia.
- U tehotných žien s fenylketonúriou môže diéta, ktorá nie je striktne dodržaná, viesť k potratu (spontánny potrat).
- V každom prípade by tehotné ženy s fenylketonúriou mali vyhľadať radu a usmernenie od lekára.
Fenylketonúria: priebeh a prognóza ochorenia
Ak je fenylketonúria diagnostikovaná včas, pokiaľ je to možné u novorodenca, a ak sa dodržiava špeciálna diéta PKU, prognóza je zvyčajne dobrá. Deti sa duchovne rozvíjajú a majú priemernú dĺžku života.
Ak sa však nelieči, poškodenie mozgu vedie k vážnym poruchám duševného vývoja, ktoré nemožno neskôr napraviť. Títo postihnutí majú normálnu dĺžku života, ale ich inteligenčný kvocient je takmer vždy veľmi hlboko pod normou.
Zvláštnym prípadom je vzácna atypická forma fenylketonúrie, v ktorej je prítomný nedostatok BH4. Tento variant fenylketonúrie môže napriek diéte viesť k progresívnemu neurologickému poškodeniu s vážnymi kŕčmi.
Tagy: alternatívna medicína zdravé pracovisko športová kondícia


.jpg)