Spinálna svalová atrofia
a Florian Tiefenböck, lekár Aktualizované dňaMaximilian Reindl študoval chémiu a biochémiu na LMU v Mníchove a od decembra 2020 je členom redakčného tímu Oboznámi sa s vami s témami lekárskej, vedeckej a zdravotnej politiky, aby boli zrozumiteľné a zrozumiteľné.
Ďalšie príspevky od Maximiliana ReindlaFlorian Tiefenböck študoval humánnu medicínu na LMU Mníchov. Ku prišiel ako študent v marci 2014 a odvtedy podporuje redakčný tím lekárskymi článkami. Po získaní lekárskej licencie a praktickej práce z vnútorného lekárstva vo Fakultnej nemocnici Augsburg je od decembra 2019 stálym členom tímu a okrem iného zaisťuje lekársku kvalitu nástrojov
Ďalšie príspevky od Floriana Tiefenböcka Všetok obsahu kontrolujú lekárski novinári.
Spinálna svalová atrofia alebo skrátene SMA je zriedkavé ochorenie, pri ktorom odumierajú určité nervové bunky v mieche. Stimuly a impulzy z mozgu potom už nedosahujú miesto určenia: svaly. To spôsobuje ochabnutie svalov a paralýzu. Existujú rôzne formy SMA. Najťažšie začína v detstve. Nové liečebné postupy sľubujú trvalé zlepšenie zdravotného stavu. Prečítajte si viac o spinálnych svalových atrofiách tu.
Kódy ICD pre túto chorobu: Kódy ICD sú medzinárodne uznávané kódy pre lekárske diagnózy. Môžu sa nachádzať napríklad v listoch lekárov alebo na potvrdeniach o práceneschopnosti. G12

Stručný prehľad
- Čo je spinálna svalová atrofia? Skupina chorôb svalovej slabosti. Sú založené na smrti určitých nervových buniek v mieche, ktoré ovládajú svaly (motorické neuróny). SMA preto patria medzi choroby motorických neurónov.
- Aké formy existujú? Dedičné spinálne svalové atrofie sú väčšinou SMA s určitým genetickým defektom na chromozóme 5 (SMA asociované s 5q). Lekári rozlišujú štyri rôzne formy: SMA typ 1 - typ 4. Okrem toho existujú sporadické formy, ktorých dedičnosť nie je istá.
- Frekvencia: Zriedkavé ochorenie; zdedená SMA postihuje asi jedného novorodenca zo 7 000.
- Príznaky: svalové zášklby, progresívna svalová slabosť, ochabovanie svalov, paralýza. Prechody sa líšia v závislosti od tvaru SMA.
- Príčiny: Dedičná spinálna svalová atrofia typu 1-4 je výsledkom genetického defektu na chromozóme 5, presnejšie na géne SMN1. V dôsledku toho telu chýba špeciálny proteín, proteín SMN. Tento deficit poškodzuje motorické neuróny v mieche.
- Diagnóza: Genetické vyšetrenie na zmenu genetickej výbavy SMN, telesné vyšetrenia, elektroneurografia, elektromyografia, krvné testy (napr. CK)
- Liečba: Je možná génová substitučná terapia alebo podanie liečiva modulátorom zostrihu. Sprievodná fyzioterapia, logopédia, terapia bolesti a psychoterapia. V prípade potreby chirurgický zákrok na chrbtici. Plán liečby v závislosti od typu SMA.
- Prognóza: V prípade dedičného proximálneho SMA majú nové možnosti terapie kauzálny účinok a môžu mať pozitívny vplyv na priebeh ochorenia. Začať liečbu včas je dôležité. Liečba nie je zatiaľ dostupná pre každého pacienta. Ak sa neliečia, deti s SMA typu 1 zvyčajne zomrú počas prvých dvoch rokov. Stredná dĺžka života typu 3 a typu 4 je len ťažko alebo nie je skrátená.
Čo je spinálna svalová atrofia?
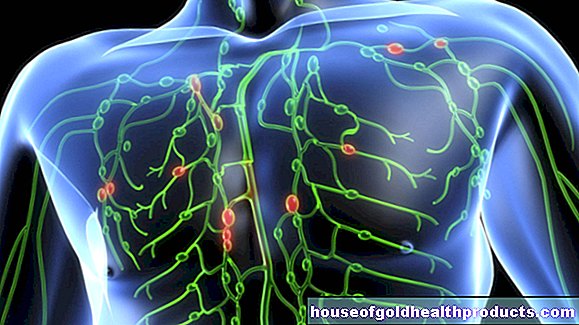
Pri spinálnej svalovej atrofii (SMA) odumierajú niektoré nervové bunky v mieche. Obvykle ovládajú svaly, a preto odborníci označujú tieto nervové bunky ako motorické neuróny. Preto SMA patria k takzvaným chorobám motorických neurónov.
Pri spinálnych svalových atrofiách sú postihnuté dolné (druhé) motorické neuróny, ktoré sú svojimi koncovkami priamo spojené so svalmi. V dôsledku poškodenia sa do svalov dostane menej alebo žiadne nervové signály. Svaly sú stále slabšie a menšie (ochabovanie svalov / svalová atrofia).
Lekári rozlišujú rôzne formy spinálnej svalovej atrofie. Jednoznačne najväčšou skupinou sú dedičné SMA, pri ktorých sú postihnuté svaly blízko kmeňa (proximálne). Sú založené na špecifickom genetickom defekte. Vyvinie ho asi jeden zo 7 000 novorodencov.
Spinálna svalová atrofia je celkovo zriedkavé ochorenie. Napriek tomu je to druhá najčastejšia autozomálne recesívna dedičná choroba. Je to tiež najčastejšia príčina smrti dojčiat alebo batoliat v dôsledku genetického defektu.
Aké typy spinálnej svalovej atrofie existujú?
Lekári rozlišujú dedičné (dedičné) formy SMA od sporadických foriem. Ďalšia klasifikácia spinálnej svalovej atrofie sa týka predovšetkým svalových skupín, ktoré sú postihnuté ako prvé. Existujú
- Proximálny SMA: S približne 90 percentami tvoria najväčšiu skupinu SMA. Príznaky začínajú vo svaloch blízko kmeňa, t.j. proximálne.
- Neximálna SMA: Tu sú najskôr postihnuté vzdialenejšie svalové skupiny, ako sú ruky a nohy (distálna SMA). V ďalšom priebehu sa tieto SMA môžu rozšíriť aj do svalov v blízkosti stredu tela.
- Špeciálne formy (napr. Spinobulbárna svalová atrofia Kennedyho typu)
Proximálne spinálne svalové atrofie
Dedičné proximálne spinálne svalové atrofie sú väčšinou ochorenia, ktoré sú založené na špecifickom genetickom defekte (SMA asociovaný s 5q, defekt na chromozóme 5). Tie sú zase rozdelené do štyroch rôznych foriem. Klasifikácia je založená na čase, keď sa objavia prvé príznaky, a na priebehu ochorenia.
Spinálna svalová atrofia typu 1: Toto je najbežnejšia a najzávažnejšia forma SMA. Hovorí sa mu aj „Werdnig-Hoffmannova choroba“ alebo „akútna infantilná SMA“. Ochorenie sa zvyčajne začína v ranom detstve. Svalová slabosť postihuje celé telo - lekári hovoria aj o „syndróme floppy infant“ (z angličtiny floppy = flaccid, infant = nemluvňa, dieťa). Väčšina neliečených detí s SMA typu 1 zomiera skôr, ako majú dva roky.
Spinálna svalová atrofia typu 2: Táto forma SMA je známa aj ako „intermediárna spinálna svalová atrofia“ alebo „chronická infantilná SMA“. Prvé príznaky sa zvyčajne objavia pred dosiahnutím veku 18 mesiacov. Títo postihnutí majú niekedy výrazne zníženú dĺžku života.
Spinálna svalová atrofia typu 3: Je známa aj ako „juvenilná spinálna svalová atrofia“ alebo „Kugelbergova-Welanderova choroba“. Táto SMA sa zvyčajne začína po dosiahnutí veku 18 mesiacov a pred ranou dospelosťou. Svalová slabosť je miernejšia ako u typu 1 alebo 2. Postihnutí majú len mierne zníženú dĺžku života.
Spinálna svalová atrofia typu 4: Je podobná SMA typu 3, ale objavuje sa iba v dospelosti (zvyčajne> 30 rokov). Svalová slabosť je však menej výrazná a postupuje pomalšie ako pri SMA typu 3.
Prechody medzi rôznymi verziami sú plynulé. V niektorých prípadoch to sťažuje jasné vymedzenie. Niektoré genetické predispozície tiež hrajú dôležitú úlohu v závažnosti príslušnej choroby.
Ostatné spinálne svalové atrofie
Okrem týchto proximálnych foriem existujú aj ďalšie formy spinálnej svalovej atrofie. Patria sem napríklad vzácnejšie, tiež dedičné distálne spinálne svalové atrofie. S nimi príznaky zvyčajne začínajú u svalových skupín, ktoré sú ďalej od tela.
Dedičnosť nie je zaručená v prípade sporadického SMA. Navyše nie je možné určiť žiadnu rodinnú akumuláciu. V literatúre patria tieto:
- Typ Hirayama (juvenilný distálny SMA, choroba okolo 15 rokov, postihuje svaly paží, zvyčajne sa zastaví aj bez terapie a môže sa dokonca zlepšiť)
- Vulpian-Bernhardov typ (tiež syndróm „flail-arm“ so začiatkom v ramennom pletenci, zvyčajne od 40 rokov)
- Typ Duchenne-Aran (spočiatku postihnuté svaly rúk, šíriace sa smerom k trupu tela, zvyčajne po 30-tke)
- Peroneálny typ (syndróm „bičenej nohy“, najskôr na svaloch dolných končatín)
- Progresívna bulbárna obrna (poruchy reči a prehĺtania, postihuje asi 20 percent pacientov s amyotrofickou laterálnou sklerózou)
Niektoré sporadické formy SMA (syndróm „flail -arm - / - noha“, progresívna bulbárna paralýza) sa v odborných kruhoch počítajú medzi varianty amyotrofickej laterálnej sklerózy (ALS). Tento článok sa zaoberá predovšetkým dedičnými proximálnymi spinálnymi svalovými atrofiami.
Spinobulbárna svalová atrofia
Spinobulbárna alebo bulbospinálna svalová atrofia (Kennedyho typ, Kennedyho syndróm) je dedičné ochorenie. Začína sa často v mladej až strednej dospelosti. Táto špeciálna forma SMA je zdedená recesívnym spôsobom spojeným s X a preto postihuje iba mužov (keďže muži majú iba jeden chromozóm X, u žien prevláda druhý, zdravý chromozóm X, ktorý by kompenzoval defekt).
Častá je svalová slabosť v svaloch blízko tela na nohách a rukách alebo ramenách, ako aj svaly jazyka a hrdla. V dôsledku toho majú postihnutí napríklad problémy s rozprávaním a prehĺtaním. Sťažujú sa tiež na chvenie, svalové kŕče a zášklby. Postihnutí muži majú tiež často zakrpatené semenníky a sú sterilní. Okrem toho sa prsné žľazy zväčšujú (gynekomastia).
Spinobulbárna svalová atrofia je zvyčajne pomalá. Stredná dĺžka života je sotva obmedzená.
Ako rozoznáte spinálnu svalovú atrofiu?
Typické pre spinálnu svalovú atrofiu sú progresívna svalová slabosť až paralýza (paréza) a svalové zášklby. V dôsledku poškodenia nervov svaly už neprijímajú elektrické impulzy, čo ich časom zmenšuje (svalová atrofia). Presné znaky a sťažnosti závisia od príslušného formulára. Nasledujúca časť sa zaoberá príznakmi dedičného proximálneho SMA.
Príznaky detskej spinálnej svalovej atrofie typu 1
Pri SMA typu 1 sa symptómy objavia v prvých šiestich mesiacoch života. Vyskytuje sa generalizovaná svalová slabosť - teda taká, ktorá postihuje celé telo. Navyše napätie medzi svalmi klesá. Lekári hovoria o hypotónii.
U novorodencov sa táto svalová slabosť spočiatku prejavuje typickým držaním nôh, ktoré pripomína ležiacu žabu (držanie žabej nohy). Nohy sú pokrčené, kolená smerujú von a chodidlá smerujú dovnútra. Dokonca ani zdvíhanie alebo držanie hlavy na vlastnú päsť zvyčajne nie je možné.
V pokročilom veku deti s SMA typu 1 nedokážu samy sedieť ani chodiť. Mnoho detí tiež nevie hovoriť, pretože môžu byť postihnuté aj svaly jazyka.
Ďalšou charakteristikou spinálnej svalovej atrofie typu 1 je tvar hornej časti tela: svaly hrudníka a chrbta sa nevyvíjajú správne. To dáva hornej časti tela zvonovitý tvar (zvonová truhlica). Vzhľadom na nízky vývoj svalov na hrudníku a chrbte zaujímajú postihnuté zhrbený postoj.
Často sa vyskytuje aj zvyšujúce sa zakrivenie chrbtice (skolióza). Zhrbený vpred a zhrbený postoj spôsobuje ďalšie problémy s dýchaním. Charakteristické je veľmi rýchle a plytké dýchanie (tachypnoe).
Príznaky strednej spinálnej svalovej atrofie typu 2
Spinálna svalová atrofia typu 2 zvyčajne spôsobuje príznaky iba vo veku od sedem do 18 mesiacov. Postihnuté deti môžu sedieť samy, ale väčšinou sa nenaučia stáť ani chodiť. Svalová slabosť postupuje pomalšie ako pri type 1.
Pri SMA typu 2 časom vznikajú symptómy podobné ťažkej infantilnej forme, ako je deformácia chrbtice. Kĺby tuhnú v dôsledku skrátených svalov a šliach (kontraktúry). Medzi ďalšie príznaky patrí chvenie rúk a zášklby svalov v jazyku.
Príznaky juvenilnej spinálnej svalovej atrofie typu 3
Spinálna svalová atrofia typu 3 sa zvyčajne objavuje po 18. roku života a pred 18. rokom života. Postihnuté deti môžu samostatne sedieť, stáť a chodiť. Svalová slabosť, najmä v svaloch panvy a nôh, však spôsobuje kolísavú chôdzu.
V priebehu niekoľkých rokov sa výkonnosť znižuje: Postihnutým sa najskôr ťažko športujú alebo lezú po schodoch, ale napokon je ťažké nosiť napríklad aj nákupné tašky. Po mnohých rokoch spinálna svalová atrofia typu 3 sťažuje alebo dokonca znemožňuje beh a akúkoľvek inú námahu, dokonca aj u starších ľudí.
Celkovo sú však symptómy menej výrazné ako u dvoch ďalších foriem chorôb, typu 1 a typu 2. U mnohých postihnutých je kvalita života po dlhšiu dobu len ťažko narušená.
Príznaky svalovej atrofie chrbtice dospelých typu 4
Táto veľmi zriedkavá forma postupného ubúdania svalov začína v dospelosti, často od tretej dekády života. Na začiatku sú postihnuté svaly nôh a bokov. Ako choroba postupuje, svalová slabosť sa šíri aj do ramien a paží.
Manifestácia klinického obrazu je podobná ako u mladistvého SMA typu 3. Progresívna svalová slabosť je však ešte pomalšia ako u SMA typu 3.
Čo spôsobuje spinálnu svalovú atrofiu?
Pri spinálnej svalovej atrofii zahynú druhé motorické neuróny v mieche. Ide o nervové bunky, ktoré svojimi prílohami ovládajú svaly. V dôsledku poškodenia týchto vysoko špecializovaných motorických neurónov sa do svalov dostane menej elektrických signálov, ako je tomu u zdravých ľudí. Ak sa svalové bunky používajú menej a teda aj menej stimulovane, telo ich časom rozloží.
Genetická chyba
Spinálna svalová atrofia je vo väčšine prípadov dedičné ochorenie (dedičná SMA). Príčinou typických proximálnych foriem SMA sú nesprávne informácie v genetickom zložení pacienta. Takzvaný gén SMN1 na chromozóme 5 nie je funkčný.
Gén SMN1 nesie informácie - tj. Plán - molekuly vitálneho proteínu nazývanej SMN. SMN znamená „Survival (of) Motor Neuron“. Bez molekuly proteínu SMN motorické neuróny časom hynú.
Je pravda, že v tele je aj príbuzný gén SMN2, ktorý je v zásade schopný „kompenzovať“ nefunkčnú genetickú informáciu SMN1. Ale to sa zvyčajne stáva len v malej miere. To znamená, že stratu funkcie génu SMN1 (ak sa nelieči) nemožno zvyčajne úplne kompenzovať intaktnou kópiou génu SMN2.
Autozomálne recesívna a autozomálne dominantná dedičnosť
Genetické informácie o osobe sú k dispozícii v dvoch vyhotoveniach. Výsledkom je, že každý má dve kópie génu SMN1 - jednu od svojho otca a jednu od svojej matky. Proximálne spinálne svalové atrofie v detstve sú typicky zdedené ako autozomálne recesívny znak.
To znamená, že oba génové varianty (alely) od rodičov musia byť defektné, aby sa u potomstva vyvinula spinálna svalová atrofia. V prípade recesívnej dedičnosti nie sú rodičia postihnutí, pretože okrem nefunkčného majú aj zdravý gén SMN1, ktorý defekt kompenzuje.
Asi každý 45. človek je majiteľom tohto systému pre SMA. Pár, v ktorom sú obaja partneri prenášačmi, má 25% riziko, že bude mať dieťa s touto chorobou.
V niekoľkých prípadoch v dospievaní nasledujú po autozomálne dominantnej dedičnosti najmä spinálne svalové atrofie v dospelosti. V prípade dominantnej dedičnosti sa defektný gén už presadí - a postihnutí ochorejú. Nejde však o vyššie uvedený genetický defekt na chromozóme 5. Tieto SMA súvisiace s 5q sú vždy dedičné autozomálne recesívnym spôsobom.
Dedičstvo s inými formulármi SMA
Dedičná môže byť aj proximálna spinálna svalová atrofia. Špeciálna spinobulbarová forma (Kennedyho typ) sa recesívne dedí prostredníctvom pohlavného chromozómu, chromozómu X (to ovplyvňuje varianty génov, ktoré obsahujú plán ukotvovacích miest pre mužské pohlavné hormóny). V prípade sporadických foriem však dedičnosť nie je zaručená. Je tu málo známe, prečo práve druhé motorické neuróny zahynú.
Vyšetrenia a diagnostika
Diagnózu spinálnej svalovej atrofie zvyčajne robia pediatri, pediatri, ktorí sa špecializujú na nervové choroby (neuropediatri) a špecialisti na choroby nervového systému (neurológovia). Na presnejšie objasnenie sú potrebné rôzne vyšetrenia. V prípade SMA sú obzvlášť dôležité genetické testy a vyšetrenia nervov a svalov.
Zbierka anamnézy (anamnéza)
Pri každej chorobe sa lekár najskôr pýta na príznaky, ktoré sa vyskytli a ako doteraz postupoval. U dojčiat a batoliat hlásia rodičia zmeny a odchýlky v správaní svojho dieťaťa. Najmä v prípade dedičných chorôb sa lekári zameriavajú aj na rodinnú anamnézu.
Fyzikálne vyšetrenia
V zásade lekár určuje abnormality v motorickom vývoji fyzickým vyšetrením dieťaťa. Testuje napríklad, či deti môžu samostatne držať hlavy vzpriamene, sedieť alebo nezávisle pohybovať (v závislosti od veku) rukami alebo nohami.
Doplnkové cvičebné testy sa vykonávajú u starších detí a dospelých s podozrením na spinálnu svalovú atrofiu. Lekár skontroluje, koľko sily môže dotknutá osoba zhromaždiť a ako dlho ju dokáže udržať. Skúma tiež vytrvalosť.
Lekár navyše testuje reflexy, ktoré sú zvyčajne oslabené alebo vyhasnuté, najmä v prípade výrazných spinálnych svalových atrofií. Za týmto účelom poklepe kladivom na rôzne šľachy, napríklad na päte alebo pod kolenom, a kontroluje reakciu.
Genetické štúdie
Najspoľahlivejšou metódou detekcie (dedičnej) spinálnej svalovej atrofie je genetická analýza. Lekári hľadajú dôkaz o zmenenom (mutovanom) géne SMN1 a počte existujúcich kópií SMN2.
Všeobecným pravidlom je diagnostikovať a liečiť (dedičné) SMA čo najskôr. V závislosti od formy a dostupnej liečby môže byť motorický vývoj pozitívne ovplyvnený skôr, ako sú motorické neuróny miechy nevratne poškodené.
Ďalšie vyšetrovanie v SMA
Ak existuje podozrenie na SMA, lekári často merajú rýchlosť vedenia nervov (elektroneurografia) a svalovú aktivitu (elektromyografia). V prípade potreby vyšetria svaly aj pomocou ultrazvuku (myosonografia) alebo magnetickej rezonancie (MRI).
Lekári navyše zabezpečujú krvné testy. Ak existuje spinálna svalová atrofia, niektoré parametre je možné zmeniť: napríklad sa zvýši hladina kreatínkinázy (CK, typický svalový enzým).
Liečba spinálnych svalových atrofií
Liečba spinálnej svalovej atrofie je komplexná. Kauzálna terapia nebola dlho možná pre žiadnu formu SMA. Pokroky v lekárskom výskume však poskytujú lekárom nové možnosti liečby, ktoré zásadne pomôžu postihnutým s proximálnym SMA (defekt génu SMN na chromozóme 5).
Lekári sa navyše zameriavajú na zmiernenie symptómov a poskytnutie najlepšej možnej podpory postihnutým (napr. Fyzioterapia, respiračná terapia, psychoterapia, prípadne chirurgický zákrok).
Lekárska terapia
Nové liečebné prístupy pre pacientov, u ktorých je SMA založená na známom defekte génu SMN, zasahujú priamo do samotného genetického materiálu alebo do následného spracovania genetickej informácie.
Cieľom je umožniť telu pacienta nezávisle produkovať dostatočné množstvo proteínu SMN, ktorý je zásadný pre motorické neuróny.
Na liečbu spinálnej svalovej atrofie sú k dispozícii nasledujúce možnosti:
- Spojovacie modulátory (Nusinersen, Risdiplam): Tieto lieky zasahujú priamo do ďalšieho spracovania molekúl messengerovej RNA. Posilňujú tie procesy, ktoré dodávajú vyššie množstvo proteínu SMN z neporušeného génu SMN2.
- Génová substitučná terapia (Onasemnogene Abeparvovec): Táto terapia zasahuje priamo do ľudského genómu. Chybná kópia génu SMN1 je v postihnutých bunkách nahradená externe dodávaným funkčným génovým konštruktom.
Spojovacie modulátory
V prípade defektu génu SMN1 môže telo alternatívne produkovať proteín SMN zo súvisiaceho génu SMN2. Náhradný gén SMN2 „naskočí“, ale to nestačí. Dôvod: Proteíny v SMN2 sú zvyčajne príliš krátke a rýchlo sa rozkladajú.
Je to spôsobené spracovaním zodpovedajúcej mediátorovej RNA SMN2 (mRNA SMN2). Prenáša stavebné informácie z genómu (DNA) do miest produkcie proteínov (ribozómy).
Za týmto účelom sa najskôr číta gen SMN2 v genóme. Vyrába sa predbežná SMN2 messengerová RNA. Okrem iného sa musí ďalej spracovávať takzvaným spájaním. Až potom vzniká zrelá messengerová RNA. Špeciálne bunkové komplexy, ribozómy, potom čítajú zrelú messengerovú RNA a tak produkujú proteín SMN2. A práve to je skrátené a nestabilné, rýchlo sa demontuje a nemôže prevziať funkciu SMN1.
Aby sa to zmenilo, účinné látky Nusinersen a Risdiplam ovplyvňujú ďalšie spracovanie predbežnej mediátorovej RNA. Výsledkom je, že tieto takzvané modulátory zostrihu v konečnom dôsledku zvyšujú množstvo použiteľných proteínov SMN - a môžu tak zaistiť adekvátny prísun.
Nusinersen
Liečivo Nusinersen je takzvaný „antisense oligonukleotid“ (ASO). Bol schválený Európskou agentúrou pre lieky v roku 2017. ASO sú umelo vyrobené a špeciálne upravené molekuly RNA. Špecificky a presne sa viažu na mediátorovú RNA SMN2. To zabraňuje ich ďalšiemu nesprávnemu spracovaniu v ľudskej bunke.
Konkrétne: Nusinersen zabraňuje tomu, aby boli dôležité informácie (exón 7) chybne vystrihnuté z mediátorovej RNA SMN2. Miesto exónu 7 spôsobuje, že telo si následne vyrobí funkčnejší proteín SMN.
Nusinersen sa podáva prostredníctvom bedrovej punkcie. To znamená, že liečivo sa vstrekuje do miechového kanála injekčnou striekačkou. Táto terapia sa opakuje v pravidelných intervaloch niekoľkých mesiacov. V prvom roku liečby dostanú postihnutí šesť, potom tri dávky ročne.
Pacienti liek zvyčajne dobre znášajú. Nusinersen vedie k priaznivejším kurzom chorôb. Štúdie ukázali, že u mnohých pacientov sa zlepšuje pohyblivosť: v mnohých prípadoch bolo možné voľne sedieť a nezávisle otáčať telo. Vedľajšie účinky a komplikácie sú okrem iného založené na lumbálnej punkcii (napr. Bolesť hlavy, infekcie mozgových blán).
Risdiplam
Európska komisia schválila v marci 2021 Risdiplam ako tretí liek proti SMA spojenému s 5q (typy 1-3 alebo jedna až štyri kópie génu SMN2). Risdiplam sa užíva denne ako rozpustený prášok. Presná dávka sa vypočíta na základe veku a telesnej hmotnosti.
Na rozdiel od Nusinersena nie je Risdiplam „antisense oligonukleotid“, ale malá molekula. Táto molekula sa viaže na mediátorovú RNA pre proteíny SMN2 a stabilizuje ich týmto spôsobom. V dôsledku toho sa vytvorí viac funkčných proteínov SMN.
Bežné vedľajšie účinky lieku Risdiplam zahŕňajú gastrointestinálne ťažkosti, vyrážky, horúčku a infekcie močových ciest.
Génová substitučná terapia
Ďalší prístup k liečbe proximálnej spinálnej svalovej atrofie sa opiera o takzvanú génovú substitučnú terapiu. Defektný gén SMN1 - východiskový bod progresívneho SMA - je „nahradený“ novou funkčnou kópiou génu.
Účinná látka Onasemnogene Abeparvovec (AVXS-101), ktorá funguje na tomto princípe, dostala od Európskej agentúry pre lieky (EMA) v máji 2020 podmienečné povolenie na uvedenie na trh liečby batoliat a detí.
Podľa informácií EMA je možné liek použiť na SMA typu 1. Vo všetkých ostatných formách ochorenia SMA rozhodujú o tom, či je alternatívou génová substitučná terapia, genetické vlastnosti (počet kópií SMN2).
Pri Onasemnogene Abeparvovec je funkčná kópia ľudského génu SMN1 zavedená do postihnutých buniek miechy a mozgového kmeňa. Vykonávajú to niektoré vírusy, ktoré slúžia ako „trajekty“ pre nový genetický materiál-takzvané adeno-asociované vírusové vektory (AAV vektory).
Vektorové génové konštrukty sa podajú raz ako infúzia cez žilu do krvného riečišťa a odtiaľ sa distribuujú do celého tela. Vzhľadom na ešte nie celkom vyvinutú hematoencefalickú bariéru u malých detí sa tieto vektory môžu dostať aj do tkaniva miechy.
Prednostnou väzbou týchto vektorov na špeciálne povrchové štruktúry motorických neurónov prednostne zaberajú genetický materiál, aby potom mohli nezávisle produkovať proteín SMN.
Liečba môže zlepšiť motorické funkcie a viesť k trvalému vývojovému úspechu (napr. Sedenie, plazenie a chôdza bez opory).Počas liečby sa niekedy môžu hodnoty pečene výrazne zvýšiť, ale počet krvných doštičiek môže klesnúť. Častá je aj horúčka a zvracanie. Na zníženie vedľajších účinkov dostávajú pacienti niekoľko týždňov kortikosteroidy („kortizón“).
Motorický vývoj primeraný veku je spravidla možný iba vtedy, ak sa génová terapia začala presymptomaticky. Liečba prebieha v špecializovaných neuromuskulárnych liečebných centrách.
fyzická terapia
Fyzioterapia je aj naďalej dôležitým pilierom liečby SMA. Nie všetky formy SMA je možné liečiť novými liečebnými prístupmi. Pravidelná cvičebná terapia je navrhnutá tak, aby udržiavala fyzické schopnosti a spomaľovala odbúravanie svalov.
Fyzioterapeut pasívne pohybuje časťami tela, ktoré sú už paralyzované. Aktívne pohybové sekvencie sú zasa trénované tak, aby podporovali pohyblivosť a silu svalov. Pomôcť môže aj masáž alebo tepelné a studené kúry. Tieto slúžia aj na relaxáciu a za určitých okolností spomaľujú ďalšiu degeneráciu.
Logopédia
V niektorých prípadoch SMA ovplyvňuje hovoriace a prehĺtacie svaly. Potom pomôže logopedické cvičenie. Povzbudzuje deti, aby sa naučili hovoriť. Aj u starších pacientov to môže zvyčajne spomaliť zhoršenie reči. Logopédi trénujú aj správne prehĺtanie.
Fyzioterapeuti aj logopédi podporujú postihnutých cielenou dychovou terapiou.
Liečba zmierňujúca bolesť
Terapia bolesti hrá dôležitú úlohu, najmä v pokročilejších štádiách ochorenia. Lekári používajú lieky zmierňujúce bolesť, aby znížili utrpenie postihnutých.
chirurgia
Pretože spinálna svalová atrofia môže viesť k závažnému zakriveniu chrbtice (skolióza), lekári niekedy zvažujú chirurgický zákrok. Pritom konkrétne spevňujú chrbticu.
Postihnutým to poskytuje (istú) dodatočnú stabilitu trupu, ktorá umožňuje nielen vzpriamenejšie držanie tela, ale tiež chráni kosti a kĺby. Operácia chrbtice môže tiež pomôcť proti progresívnym problémom s dýchaním.
Psychoterapeutická starostlivosť
Neuromuskulárne ochorenia, ako je spinálna svalová atrofia, predstavujú veľký psychický stres. Pacienti a príbuzní spracúvajú diagnózu na individuálnych a skupinových sedeniach vedených psychoterapiou a vyvíjajú stratégie na lepšie zvládnutie choroby.
Svojpomocné skupiny a zástupcovia pacientov tiež ponúkajú dôležitú podporu. Informujú, radia a podporujú postihnutých a ich príbuzných, aby zvládli výzvy súvisiace s chorobou SMA.
Šance na zotavenie zo spinálnych svalových atrofií
Ak existuje spinálna svalová atrofia, prognóza závisí predovšetkým od príslušného tvaru. Čím neskôr sa príznaky objavia, tým lepší bude priebeh. Navyše, čím skôr lekári diagnostikujú spinálnu svalovú atrofiu, tým skôr môžu začať vhodné liečebné opatrenia, dokonca aj predtým, ako sú motorické neuróny nenávratne poškodené.
Nové možnosti liečby prostredníctvom modulátorov zostrihu a génovej substitučnej terapie majú veľký potenciál pri liečbe proximálneho SMA - najmä pri začatí liečby (veľmi) skoro. Údaje o spoľahlivej dlhodobej prognóze však stále nie sú k dispozícii. Iba ďalšie štúdie a dôsledné pozorovania bezpečnosti liekov môžu poskytnúť ďalšiu istotu v priebehu nasledujúcich (mesiacov a) rokov. Pri novších liekoch je prinajmenšom mysliteľná dlhodobá kontrola choroby alebo dokonca vyliečenie.
SMA typu 1 je spravidla vážna choroba. Deti, u ktorých sa vyvinie SMA typu 1, majú (neliečenú) veľmi obmedzenú dĺžku života. Rýchlo rastúca svalová slabosť v celom tele ovplyvňuje aj dýchanie. Následkom je akútny zápal pľúc a dokonca respiračné zlyhanie. Postihnuté deti zomierajú v prvých rokoch života.
Prognóza je pre SMA typu 2 o niečo lepšia. Stredná dĺžka života sa líši v závislosti od presnej závažnosti ochorenia: niektorí zomierajú v detstve, ale väčšina z nich dosiahne mladú dospelosť. V ťažších formách musí byť skôr alebo neskôr - ak je to žiaduce - podporované dýchanie. Postihnuté osoby zostávajú mobilné pomocou invalidného vozíka.
Pri SMA typu 3 je prognóza výrazne lepšia - najmä ak sa prvé príznaky objavia neskoro. Výkon sa v priebehu niekoľkých rokov postupne zhoršuje. V starobe môže byť potrebný invalidný vozík alebo dokonca trvalá starostlivosť. Stredná dĺžka života je ťažko obmedzená spinálnou svalovou atrofiou typu 3.
Spinálna spinálna svalová atrofia (typ 4) je ešte pomalšia ako typ 3. Ľudia majú zvyčajne normálnu dĺžku života.
Tagy: liečivé bylinné domáce opravné prostriedky symptómy nesplnené želanie mať deti




















.jpg)